El síndrome de Marfan afecta a casi todos los órganos del cuerpo, incluyendo el corazón y la arteria aorta. No podemos, por tanto, hablar de cardiopatía congénita en adultos porque, pese a ser una enfermedad hereditaria, no se limita al corazón.
Sin embargo, los padres de un bebé afectado por el síndrome de Marfan se sienten tan perdidos como los que nos preguntan por qué nace un bebé con cardiopatía congénita y pueden experimentar también un equivocado sentimiento de culpabilidad. Hoy vamos a ver cómo proceder con la afectación del síndrome de Marfan al corazón y la aorta. Los demás órganos afectados serán tratados por los especialistas correspondientes.
¿Cómo afecta el síndrome de Marfan al corazón?
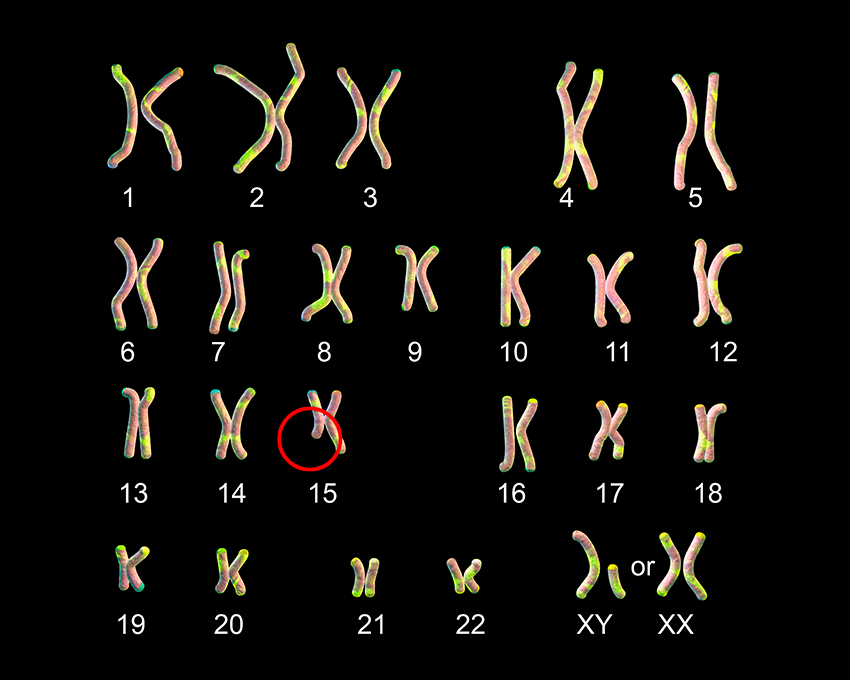
El síndrome de Marfan es una enfermedad causada por una alteración en el cromosoma 15, encargado de regular la síntesis de fibrilina. Esta proteína es indispensable en la producción del tejido conectivo, presente en todos los órganos, desde los huesos hasta la piel. Por eso no es una enfermedad cardíaca, o no solamente, aunque el corazón es uno de los órganos más afectados y determinantes en la esperanza de vida de estos pacientes, la cual se ha logrado aumentar de manera considerable.
¿Qué consecuencias tiene el síndrome de Marfan?
A medida que el paciente crece, aparecen problemas en el corazón, la vista, la columna vertebral y los pulmones. También hay otras estructuras, como son los ligamentos y la piel que pueden resultar afectadas, pero las que más preocupan son las que acabamos de mencionar.
Los pacientes con síndrome de Marfan precisan de la intervención coordinada de un cardiólogo, para controlar y reducir los daños en el corazón y la dilatación de la arteria aorta, un neumólogo, pues pueden llegar a sufrir colapso pulmonar, un traumatólogo, un oculista y un genetista.
¿Cuál es la causa?
Aceptamos que el síndrome de Marfan es una enfermedad congénita, con patrón de herencia autosómico dominante, lo que quiere decir que, estadísticamente, la probabilidad de que los hijos de un paciente estén afectados por la enfermedad es de aproximadamente un 50 %. Sin embargo, alrededor del 25 % de los pacientes no hay un progenitor con la sintomatología.
Esto quiere decir que, o bien hay otras causas no identificadas, o que tal vez el progenitor que porta el gen dañado ha presentado una de las formas más leves del síndrome y ha pasado inadvertido, aunque eso puede resultar algo extraño.
Cuando se sabe que uno de los padres es portador de esta alteración cromosómica, un estudio genético puede determinar si el bebé nacerá o no afectado, y en caso de ser así es posible proceder al control y tratamiento del síndrome Marfan antes de que produzca daños severos en el corazón y en otros órganos vitales.
Síntomas
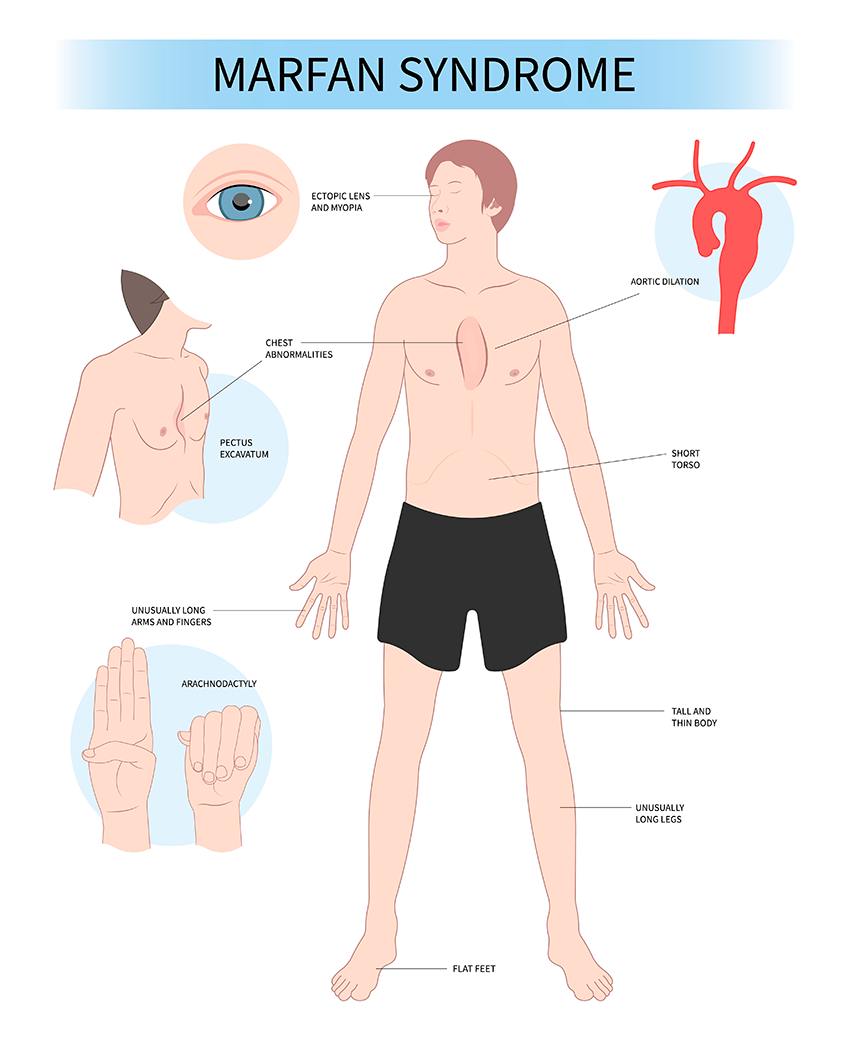
Las personas portadoras del gen responsable del síndrome de Marfan suelen ser altas y delgadas, con brazos, piernas y dedos extraordinariamente largos, pudiendo hablarse de desproporción. También son personas tremendamente flexibles por lo que respecta a sus articulaciones, aunque esto lejos de ser una ventaja puede ser un problema si se exponen a deportes intensos y, sobre todo, a deportes de contacto.
A medida que se desarrollan pueden aparecer otros síntomas, como ojos hundidos, la mandíbula pequeña, el paladar alto y arqueado, y los dientes amontonados, que se suman a un pecho anormalmente cóncavo por exceso o por defecto y a desviaciones de columna tan importantes como para requerir el uso de un corsé ortopédico.
Diagnóstico y tratamiento
Cuando no conocíamos bien el genoma humano, el diagnóstico del síndrome de Marfan solía hacerse observando a los hijos de pacientes afectados desde su nacimiento y con la exploración de los pacientes que, por su morfología, pudieran presentar esta alteración. Se realizaba un ecocardiograma y placas de tórax, para ver la estructura de la caja torácica y medir desviaciones vertebrales.
En la actualidad, aunque esas pruebas siguen siendo una ayuda para conocer el grado de afectación del paciente, con un estudio genético a partir de una muestra de sangre podemos saber si estamos ante un síndrome de Marfan o no.
Por lo que respecta al tratamiento, supondría una reparación genética que todavía no sabemos hacer. Por este motivo, lo que se hace es ir controlando a los pacientes y corrigiendo los daños importantes antes de que sean graves. Las revisiones periódicas son obligatorias, como también lo es llevar una vida relativamente tranquila.
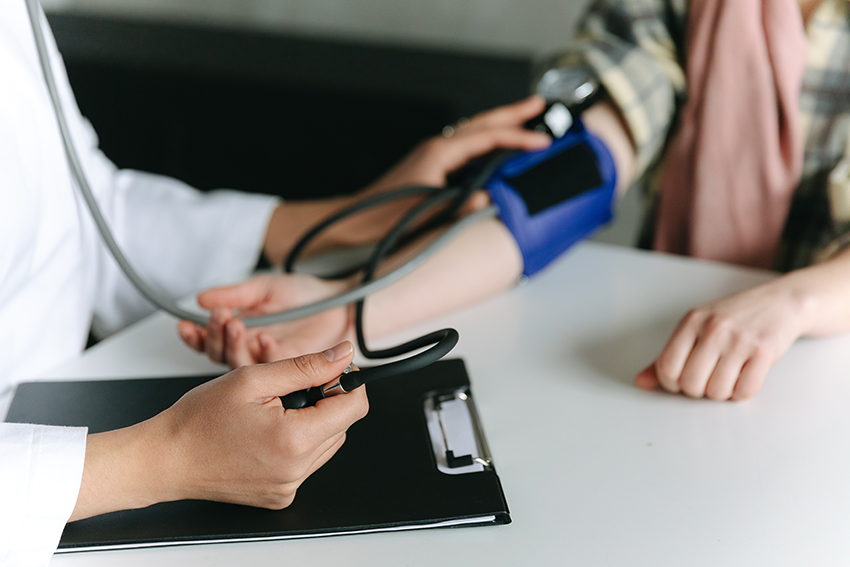
Estos pacientes deben evitar la hipertensión arterial para que no se dilate todavía más su arteria aorta, por lo que se procede a medicarles con antihipertensivos antes que a personas que no presentan este síndrome. Una dieta pobre en sodio es un buen hábito desde el nacimiento, sumado a los controles periódicos y a la fisioterapia.
En realidad, cada paciente con síndrome de Marfan necesita su propio tratamiento adaptado, a veces con correctores ortopédicos, y otras con cirugías, sumado a un estilo de vida que no suele ser muy restrictivo.
El síndrome de Marfan es un problema que afecta al tejido conectivo, el corazón y la aorta son los órganos vitales más afectados. La afectación del síndrome de Marfan al corazón y la aorta suele ser, junto con algunos casos de neumotórax, los factores más preocupantes para la supervivencia de los pacientes no diagnosticados a tiempo.
Con un componente genético, la detección precoz y el seguimiento rutinario son la mejor terapia que se puede ofrecer actualmente a estos pacientes. Con las revisiones y las correcciones necesarias, quirúrgicas y ortopédicas, se ha logrado aumentar de manera considerable la esperanza de vida de las personas afectadas en modo severo por este síndrome. Los casos leves pueden llegar a no necesitar tratamientos invasivos para prolongar la esperanza de vida, aunque sí las revisiones y un estilo de vida determinado.